7 月 19 日,开发超灵敏测序分析技术的基因组学和分子诊断公司阅尔基因与英国微软研究院在顶级学术期刊《Nature Communications》上公布了一项最新研究成果,展示了一种深度学习模型(DLM),用于从DNA探针序列预测NGS测序深度。该研究是利用机器学习提高基因组学效率和准确性的最新成果。
主要研究内容
DNA靶向测序是进行高通量基因组学和临床研究的主要方法,并且越来越多地被用于分子诊断。受限于检测技术成本及人类基因组庞大的数据量,在临床应用中对人类基因组进行深度测序,现阶段经济上可行性很低,因此在临床诊断中,通常利用杂交捕获探针来富集目标DNA区域(即靶向测序)。但用于靶向测序panel的DNA寡核苷酸探针在与其各自靶标基因位点结合时,通常遵循不同的动力学和热力学条件,因此仅仅单纯设计合成一组DNA探针将导致不同基因位点的富集效率截然不同,引起测序阶段不同基因位点之间测序深度的不均一。
而NGS技术检测某基因位点的灵敏度与该基因位点区域的测序深度是成正比的,因此针对测序深度的不均一性,需要增加额外的测序成本来保证所有基因位点都能达到最小测序深度,否则将导致临床检测灵敏度降低。依据经验来对NGS panel的探针序列和浓度等条件进行优化费时又费力,但目前别无选择。因此,能够准确预测 DNA 结合动力学的算法,将有助于改善并推进NGS panel在临床领域的发展应用。
虽然近年来DNA测序成本呈指数下降,但测序深度均一性不足会导致在高深度靶标区域浪费大量冗余reads, 而在低深度靶标区域可提供的信息却又不足。因此,合理设计高度均一NGS Panel的需求极为强烈。但对于研究者来说,即使是单重的基因位点的探针设计,预测其DNA杂交动力学效率、得到其测序深度也是极其困难的。在需要富集多重靶标的复杂体系中,预测测序深度则更加困难。
机器学习-深度学习可以利用大数据集自主探究输入和输出之间的弱相关特征。这使得深度学习可以在计算机数据可视化及其他可产生大数据集的领域占据主导地位。庞大的NGS数据集则更加适合机器学习。另一方面,目前在自然科学和生物医学领域中简单的统计模型(例如多元线性回归)仍然占主导地位,针对特定问题进行深度分析的数据很少。
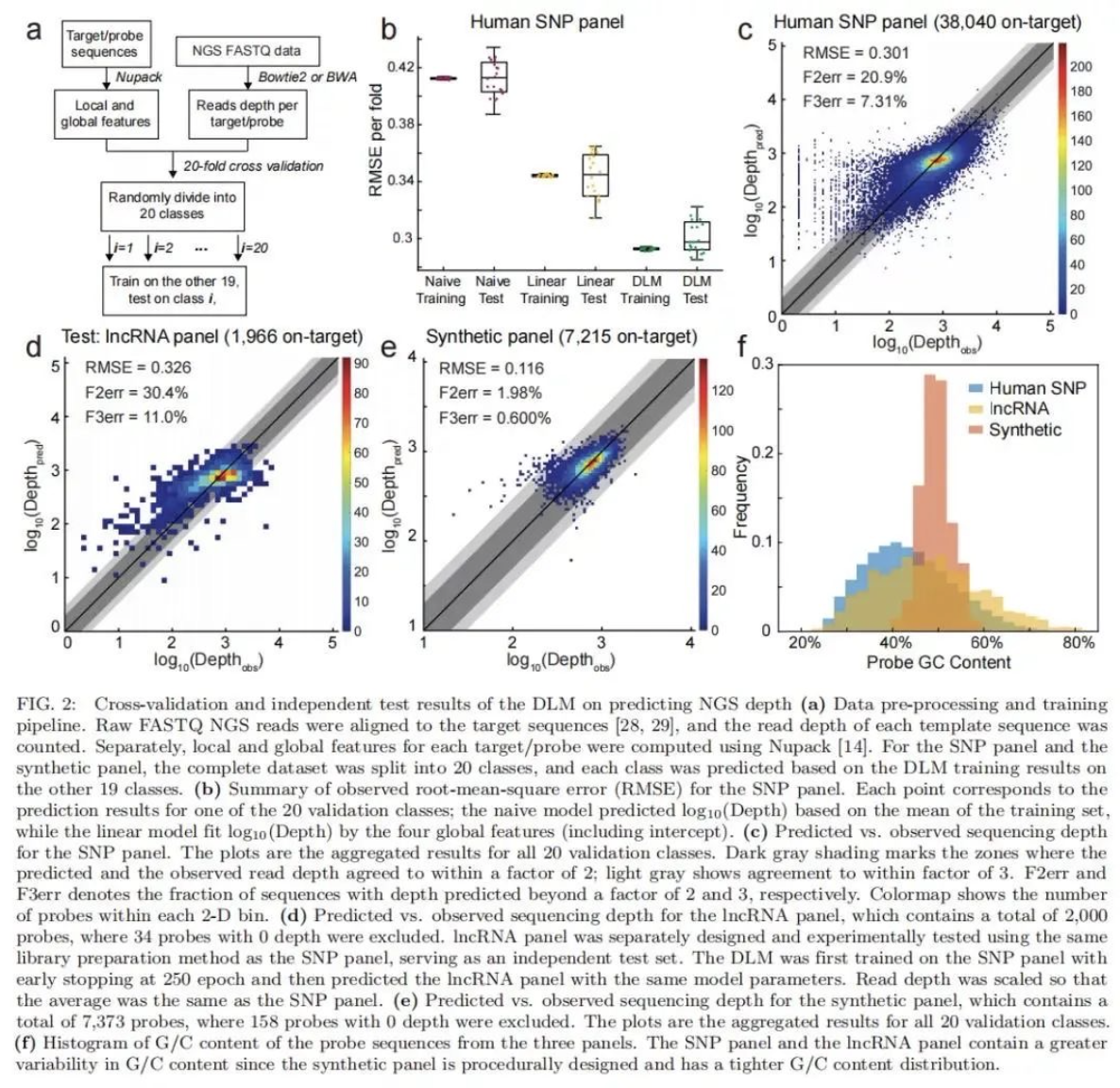
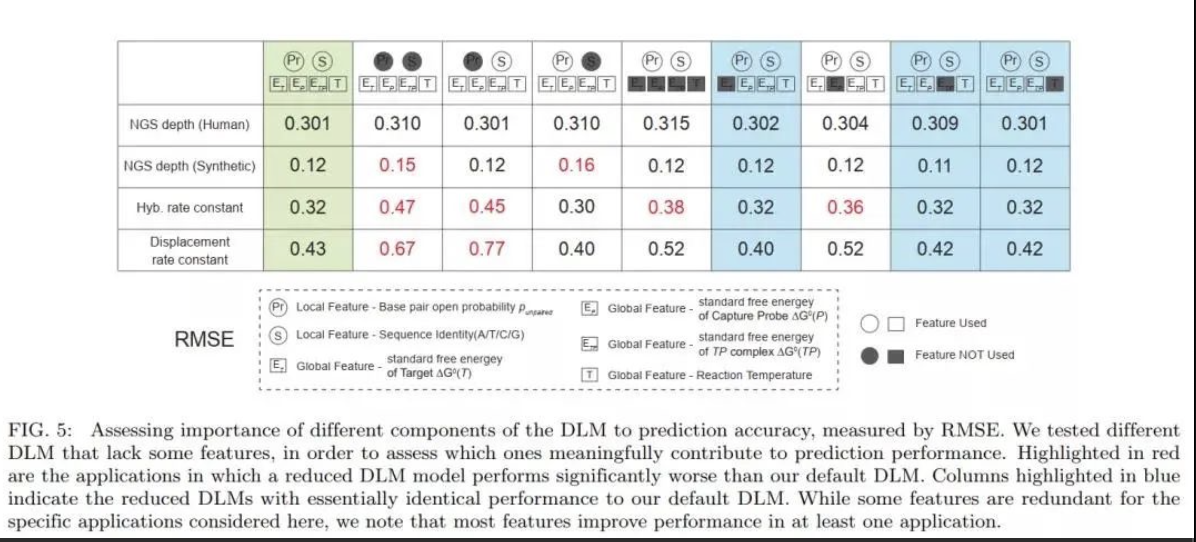
研究团队基于循环神经网络(RNN)构建了一种深度学习模型(DLM),DLM可以更好地获得 DNA 探针序列内可能影响捕获效率和捕获速度的短程及长程相互作用。该研究将DLM用于预测给定寡核苷酸探针的测序深度,并测试了其在3种NGS Panel中的性能。三种NGS panel分别为包含39145个人类基单核苷酸多态性探针的SNP panel、包含2000人类长链非编码RNA的lncRNA panel、包含7373个人工合成序列的用于存储信息的合成panel。SNP panel的测试结果表明,基于不同序列测序深度的DLM预测值(log10 Depth)均方根误差(RMSE)与实际测序后分析得到的 NGS 读取深度相结合,可消除机器学习针对噪音训练的可能性;lncRNA panel 的结果表明,DLM在兼容不同的panel时不会产生实验差异。
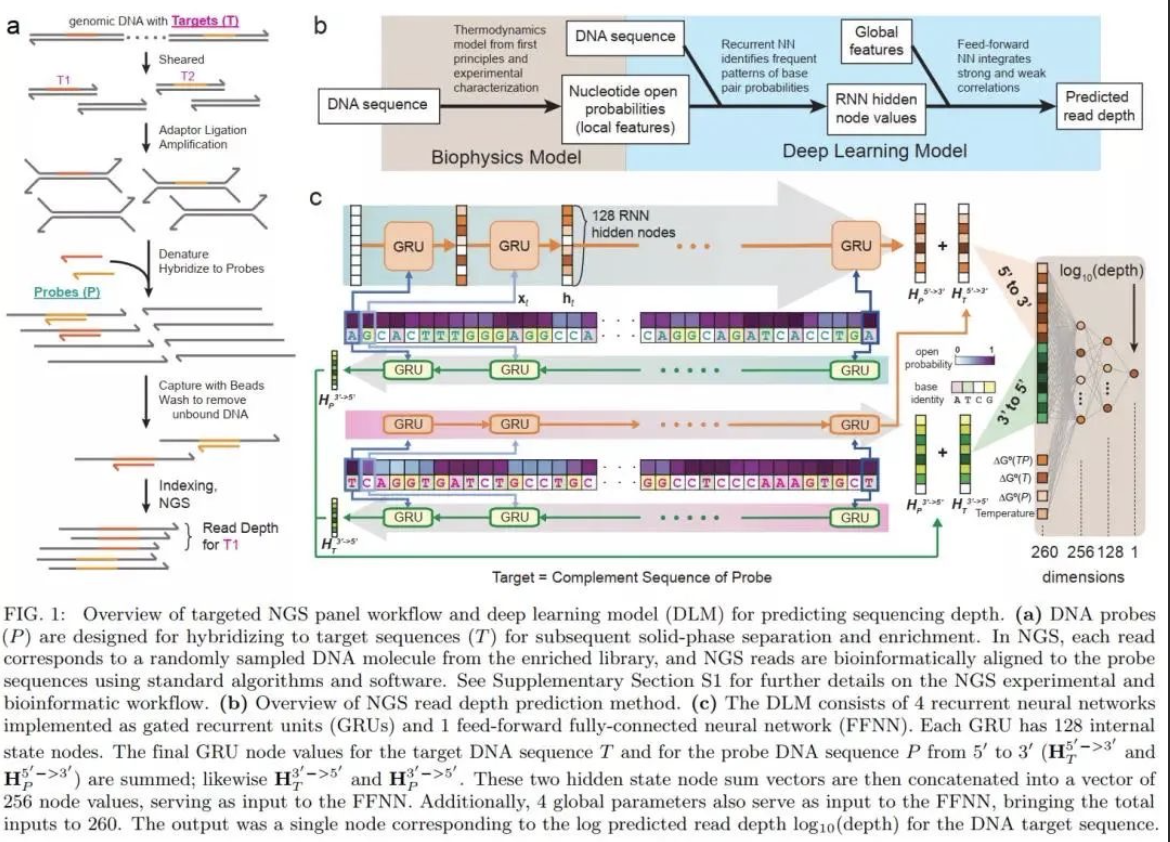
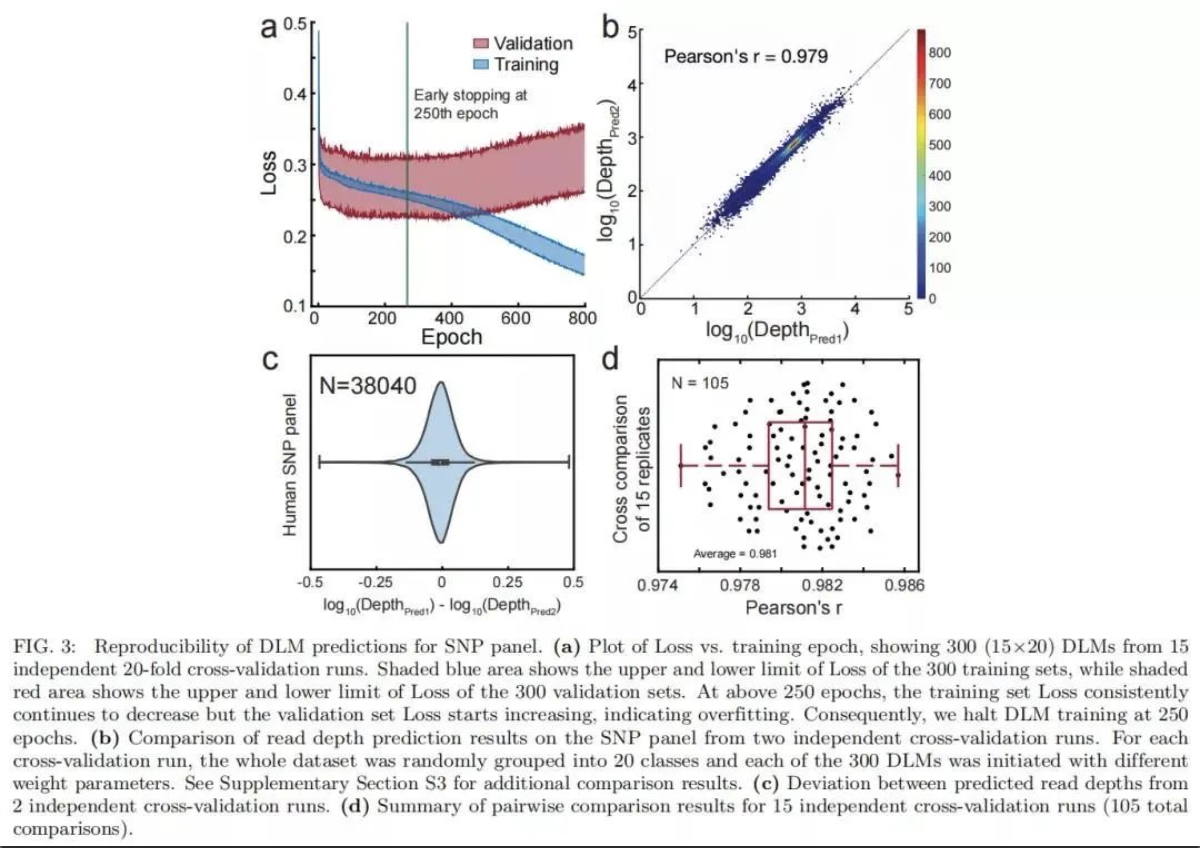
DLM 总共包含超过300,000个参数(例如节点偏差和节点-节点权重)。为了解决大量参数可能引起的过度拟合和模型重复性问题,研究团队在SNP panel测试中进行了 15次独立的20倍交叉验证,共计300 个(15×20)DLM预测的测序深度显示出高度一致性
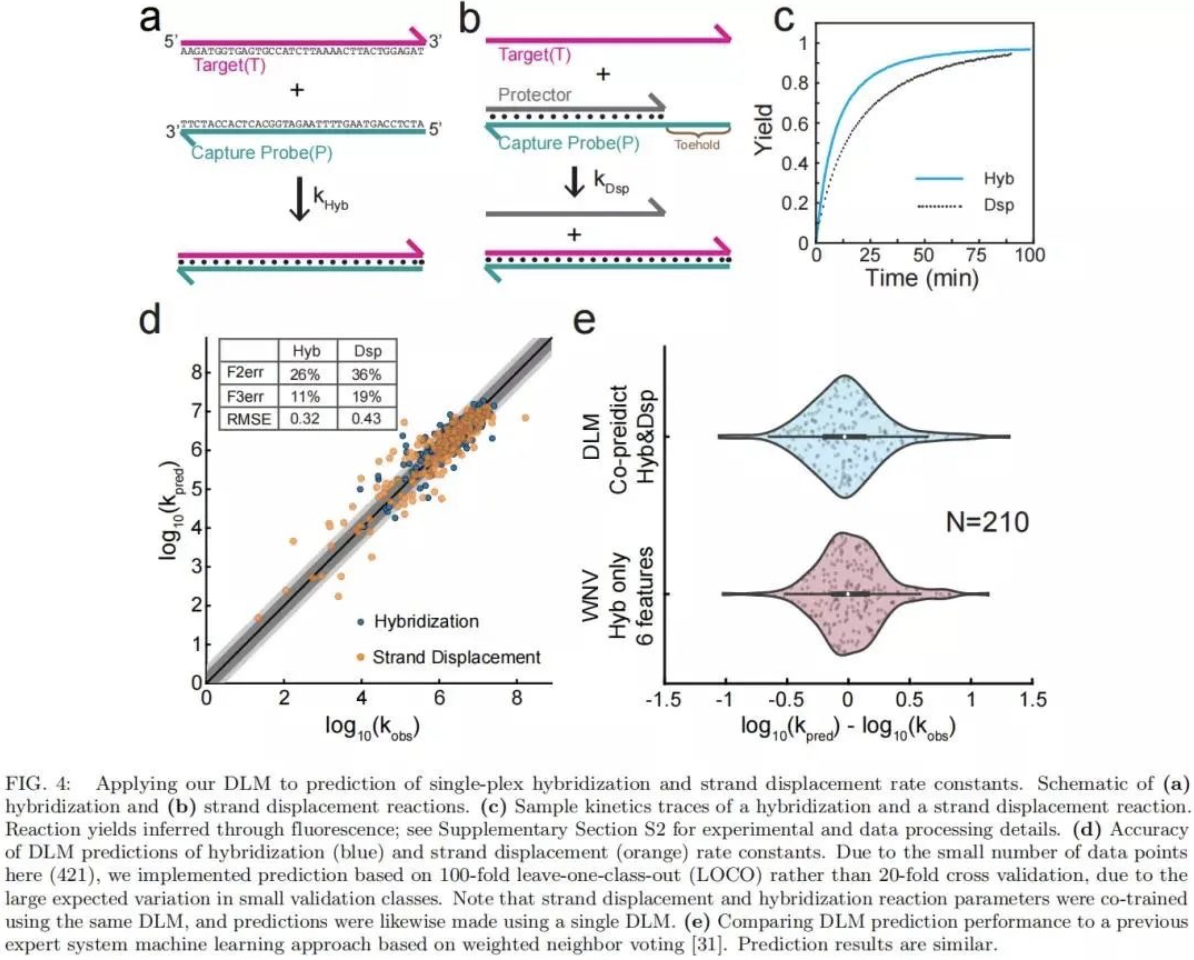
研究发现,NGS的reads数和测序深度首先取决于 DNA探针杂交的产量和速率,其次是化学特异性的偏差。 因此,该DLM在预测DNA 杂交的速率常数方面也应该是有效的(图 4a)。为了进一步推演和验证DLM方法的有效性,研究团队还进行了DLM预测DNA杂交速率常数的有效性测试,将DLM应用于DNA链置换反应的预测 ,基于实时荧光数据,观察杂交反应和链置换反应的实验最佳拟合速率常数,并将DLM对杂交率常数的预测与早期的专家系统机器学习方法进行了横向对比,其预测结果是一致的。该结果预示了DLM用于核酸分子诊断的非NGS应用(例如基于qPCR和电化学的应用)可能性。更重要的是,该DLM同时接受了杂交和链置换速率常数数据集的训练。
DNA序列与深度学习网络解决的问题中的大多数因素不同。DNA分子有长距相互作用,如远端DNA核苷酸可相互结合。此外,远端和分子间DNA结合本质上是化学效应,没有类似于自然语言处理中的有规律性的语法规则,并不需要符合人类直觉,难以通过神经网络模式发现。这些都增加了构建基于序列来预测DNA行为的神经网络的难度。
然而,一旦建立了神经网络架构来“理解”DNA 序列,就可能有潜力用于解决大量基于核酸的问题。例如在科研领域,现已发现了大量的非编码RNA,若能预测它们的结构将有助于洞悉其功能。在生物医学领域,例如可用于合成生物学的密码子优化问题,包括从头构建基于RNA的药物。考虑到核酸序列对其形式和功能,以及对应长度序列数量的影响,将可归纳的领域知识与深度学习架构结合,将成为预测核酸行为的关键推动因素。
作者点评
阅尔基因美国创新中心的高级科学家、该研究的主要作者Jinny Zhang表示:“测序均一性差的NGS panel浪费了大部分测序reads,并且对于低深度靶标区域可提供的信息不足。”
微软研究院前首席科学家、该研究的共同作者博扬·约尔丹诺夫(Boyan Yordanov)表示:“我们的工作表明,我们可以利用神经网络和机器学习的其它进展来帮助设计 NGS panel。”
该研究的数据强调了DLM如何用于预测包含超过30,000 个探针的复杂NGS panel的测序深度。DLM预测NGS测序深度的准确度可以达到93%~99%。
微软英国研究院高级首席研究员、该研究的共同通讯作者Andrew Phillips表示:“这项研究结果是我们将机器学习应用于基因组学的长期合作的一部分,微软研究院致力于计算机科学和生物学交叉领域的基础和应用研究。”
阅尔基因美国创新中心负责人、莱斯大学生物工程终身教授、该研究的共同通讯作者David Zhang表示:“NGS的进步使我们整个社会都能够采用精准医学,意味着每个患者的个性化疾病生物标志物都可用于为最佳治疗提供信息。这项合作研究标志着计算设计(computationally-designed)的NGS panel取得了进展,具有更好的灵敏度,可以改善患者的获益。”

©2021 磐霖资本保留所有权利 沪ICP备10037119号-1  沪公网安备 31011502019370号
沪公网安备 31011502019370号